Generative AI for Pharma QA: CAPA + Batch Release Drafting That Survives Part 11 (On-Prem, Multi-Site CDMO/CMO)
It’s 6:30 PM, and the batch is technically “ready.”
The client wants the release call.
Operations wants the line freed.
QA wants the record clean.
But the bottleneck isn’t a lab result or an equipment status.
It’s narrative assembly: stitching SAP batch context, deviation history, attachments, and SOP language into a CAPA plan that’s specific enough to be actionable—and a batch release memo that’s consistent, defensible, and aligned with the deviation disposition.
Multiply that across sites, clients, and a mixed regulatory footprint, and “documentation work” quietly becomes the real throughput constraint.
This is where generative AI in pharma manufacturing is often introduced, usually with the wrong promise.
GenAI can help, but only if you refuse the obvious mistake: letting a model generate uncontrolled text that drifts into a shadow QMS (Word documents, email threads, copy-paste records with no evidence linkage).
In GxP environments, faster words are not value.
Value is faster, reviewable, evidence-grounded drafts, with strict boundaries between a draft workspace and the validated system of record.
This post describes a delivery-grade pattern for on-prem GenAI that accelerates CAPA documentation and batch release memo drafting—without turning compliance into a gamble.
The uncomfortable truth: your CAPA backlog is a narrative assembly problem
In a CDMO/CMO, CAPA and batch release documentation is expensive not because of typing, but because it is cross-system and high-context.
A single CAPA package commonly pulls from:
- Deviation record fields and attachments (chronology, classification, risk rationale)
- SAP batch genealogy (materials, lots, work orders, rework loops)
- Equipment history and maintenance notes (calibration and PM context)
- QC results summaries and flags (LIMS if available, but not required)
- Controlled SOP sections, forms, and templates (version accuracy matters)
- Prior similar events (for trending and consistency)
- Client-specific requirements (the hidden “fourth regulator” in CDMO life)
What consumes senior QA time is not writing.
It is retrieving evidence, reconciling inconsistencies, and producing language that is specific, testable, and not over-committed.
Multi-site operations amplify the risk. The same event type gets described three different ways. CAPA “standard language” mutates from site to site. Records become harder to trend, harder to audit, and slower to approve.
You may already be seeing similar patterns elsewhere in pharma manufacturing. For example, AI-based inspection systems are being used to standardize packaging decisions and create traceable evidence for quality outcomes—reducing subjectivity without removing accountability.
This shift is explored in AI securing pharmaceutical packaging through standardized inspection
The lesson applies directly to QA documentation: AI creates value when it strengthens consistency and evidence linkage—not when it replaces accountable decision-making.

What GenAI can do in QA (and what it must never do)
If you want GenAI to survive Part 11 expectations, you need a hard line between draft assistance and regulated records.
Safe (and high-ROI) uses in QA documentation
- Drafting from known facts
Generate first-pass CAPA plan text or batch release memo sections from structured fields and retrieved evidence. - Summarizing and structuring
Convert long deviation narratives into clear timelines, “facts-only” summaries, and missing-information checklists. - Evidence retrieval and quoting
Pull the correct SOP sections, form language, and template clauses with document version metadata. - Consistency checks
Flag contradictions, such as “minor deviation” language paired with high-risk rationale. - Template enforcement
Ensure required sections are present and phrased consistently across sites.
Unsafe uses (don’t do these)
- Deciding root cause, product impact, batch disposition, or batch release
- Approvals, electronic signatures, or final authorization language
- Creating the final GxP record outside the validated system of record
- Writing back into QMS fields or statuses without explicit human review
Practical rule:
The assistant drafts. Humans decide. Validated systems record.
The compliance constraints you can’t hand-wave (Part 11 + mixed markets)
In mixed-market CDMO environments, two realities always apply.
The record must remain trustworthy and reconstructable
If a draft influences the official record, you must be able to explain what it was based on, who reviewed it, and how it entered the system of record.
“Draft vs record” is the safety boundary
GenAI output must live in a controlled draft workspace. Only approved content is promoted into the validated system through normal workflows.
This aligns closely with broader efforts to modernize GMP documentation using AI, where the goal is clearer, more reviewable, and audit-ready records—not automation of decisions.
For deeper context, see AI in pharmaceutical quality compliance and GMP documentation
On-prem deployment helps with data exposure, but it does not remove the need for role-based access control, client segregation, auditability of inputs, change control for models and prompts, and monitoring for output quality and drift.
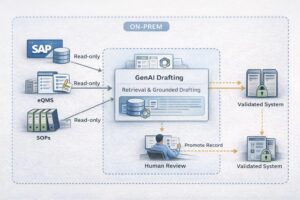
How it works: on-prem “grounded drafting” for CAPA and batch release
This architecture holds up in real QA environments because it is deliberately conservative.
1) Data sources and boundaries (read-only)
Minimum viable setup:
- SAP exports or views (batch genealogy, materials, work orders)
- Deviation and CAPA tracker exports (fields and attachments)
- Controlled SOPs and templates with version and effective-date metadata
Optional as you scale: eQMS, EDMS, MES (eBR), LIMS, training systems.
Non-negotiable rule: the assistant is read-only against all source systems.
2) Grounding pattern: retrieve first, then generate (RAG)
In regulated documentation, the model must not “know” things. It must retrieve them.
- Index controlled documents with document ID, version, effective date, owner, and applicability.
- For each drafting task, retrieve only:
- The specific record context (deviation, CAPA, batch artifacts)
- The relevant controlled document fragments
- Generate drafts with explicit evidence pointers (for example, SOP-ID, version, section).
- If evidence is missing, output “Unknown” and list what is required.
3) Tool and agent boundaries
Allowed tools
- Search controlled document index
- Fetch read-only record fields and attachments
- Populate templates
- Generate reviewer checklists
- Highlight contradictions and missing evidence
Disallowed tools
- Updating QMS fields or statuses
- Creating or closing CAPAs
- Generating signed outputs
- Sending client communications
- Attaching evidence without explicit human action
For organizations evaluating an AI data solution for pharmaceutical quality operations, this retrieve-then-generate pattern is the lowest-risk starting point.
4) Measure reviewability, not speed
Before scaling, measure:
- Grounding accuracy (claims backed by evidence)
- Citation coverage
- Hallucination rate
- Consistency across sites
- Reviewer effort and rework cycles
- Audit reconstructability
Implementation Blueprint (Multi-Site CDMO/CMO)
| Phase | Typical Timeline | Primary Objective | Key Deliverables |
|---|---|---|---|
| Phase 0: Readiness & Risk Framing | 2–4 weeks | Define safe boundaries and compliance controls | Use-case scope (CAPA + batch release drafting only), draft vs record boundary mapping, client/tenant segregation design, risk assessment and control plan, validation approach outline |
| Phase 1: Build Grounded Drafting Service (On-Prem) | 4–8 weeks | Establish controlled GenAI drafting capability | On-prem LLM runtime, controlled document index with version metadata, read-only data connectors, CAPA and release templates, baseline logging and monitoring |
| Phase 2: Pilot (Single Site / Scope) | 6–10 weeks | Prove value with measurable quality | Human-in-the-loop workflow, reviewer checklist and escalation rules, pilot evaluation report, SOP addendum for AI-assisted drafting |
| Phase 3: Multi-Site Scale & Governance | 8–16+ weeks | Standardize and govern | Harmonized templates and taxonomy, versioned prompt and policy library, governance for updates, ongoing monitoring and sampling audits |
Failure Modes and Mitigations
- Hallucinations
Mitigated through retrieve-first rules, evidence pointers, and reviewer verification. - Client data leakage
Mitigated through strict tenant isolation and scoped retrieval. - Narrative drift across sites
Mitigated through constrained templates and centrally governed prompts. - Shadow QMS behavior
Mitigated through controlled draft workspaces, promotion steps, and SOP training. - Over-trust or zero-trust
Mitigated by showing sources, gaps, and unknowns clearly.

Scenario: Batch release pressure meets CAPA documentation debt
A multi-site CDMO sees recurring packaging line deviations. Facts exist, but CAPA plan writing and batch release memo drafting slow everything down.
A grounded on-prem assistant retrieves only:
- Deviation records and attachments
- Approved templates
- Correct SOP versions
It outputs:
- A facts-only timeline with evidence pointers
- A CAPA draft that flags missing artifacts
- Effectiveness checks tied to measurable signals
QA retains full decision authority. The assistant removes retrieval friction and reduces variability.
This is for you if…
- CAPA and batch release documentation is a throughput constraint
- You require on-prem deployment and strict client segregation
- You are willing to define and enforce draft-vs-record boundaries
This is not for you if…
- You want automated CAPA closure or batch release decisions
- You want GenAI writing directly into QMS records
- You are not prepared to update SOPs, validation approaches, and training
If you are evaluating AI in pharma manufacturing, start where the risk is lowest and the return is real: evidence-grounded drafting for CAPA and batch release documentation.
As a generative AI dev company in India, Brainy Neurals focuses on delivery-grade GenAI systems that survive audits, not demos.
Request a GxP GenAI readiness assessment from Brainy Neurals to define boundaries, architecture, controls, and a pilot plan that works in real CDMO/CMO environments.
This work incorporates domain insights from Kinjal Patel, Pharma SME, focused on aligning GMP and quality requirements with practical, technology-enabled solutions.